Abstract
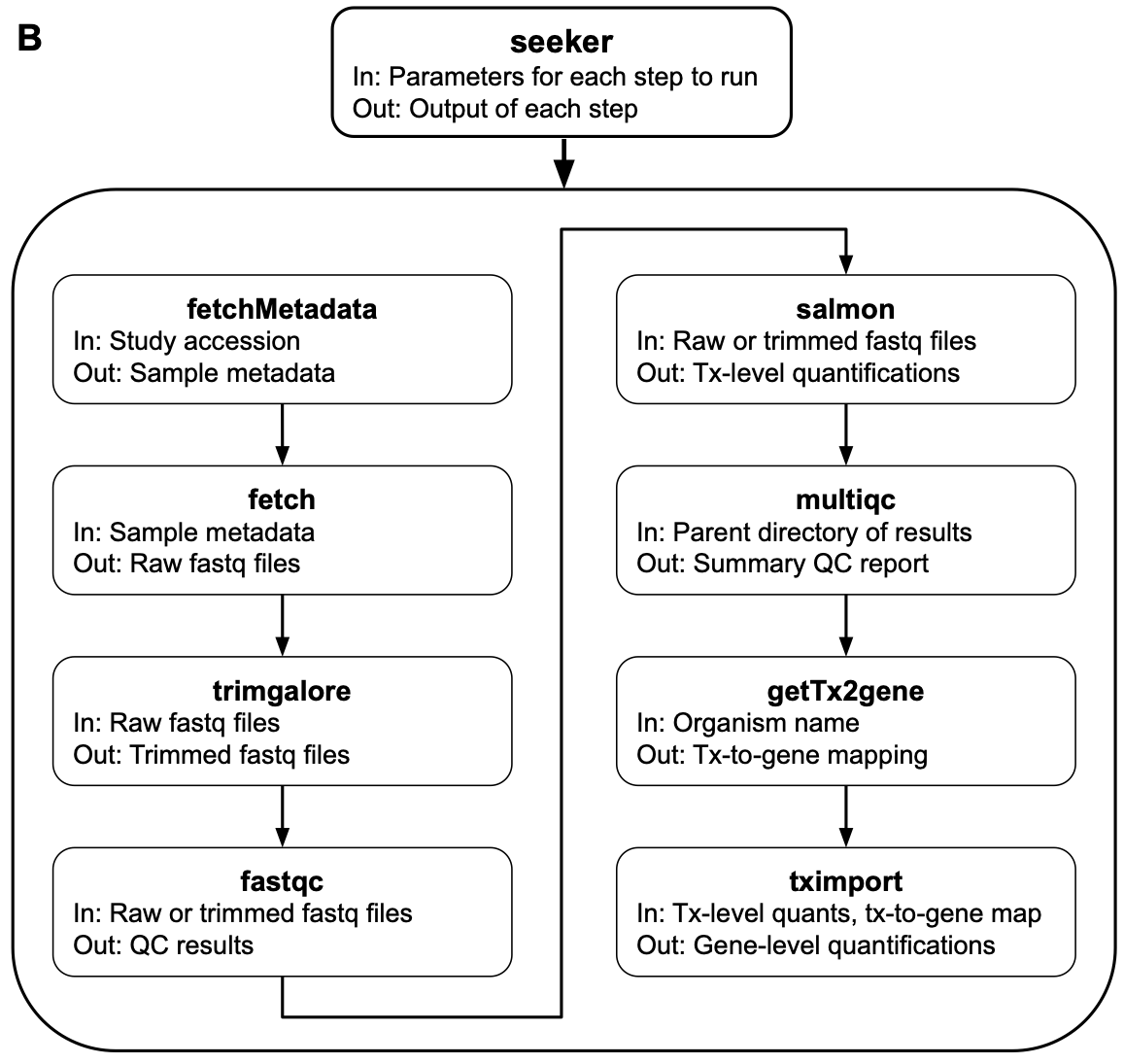
Transcriptome data have become invaluable for interrogating biological systems. Preparing a transcriptome dataset for analysis, particularly an RNA-seq dataset, entails multiple steps and software programs, each with its own command-line interface (CLI). Although these CLIs are powerful, they often require shell scripting for automation and parallelization, which can have a high learning curve, especially when the details of the CLIs vary from one tool to another. However, many individuals working with transcriptome data are already familiar with R due to the plethora and popularity of R-based tools for analyzing biological data. Thus, we developed an R package called seeker for simplified fetching and processing of RNA-seq and microarray data. Seeker is a wrapper around various existing tools, and provides a standard interface, simple parallelization, and detailed logging. Seeker’s primary output—sample metadata and gene expression values based on Entrez or Ensembl Gene IDs—can be directly plugged into a differential expression analysis. To maximize reproducibility, seeker is available as a standalone R package and in a Docker image that includes all dependencies, both of which are accessible at https://seeker.hugheylab.org.
Where applicable, full text and supplement provided for fair use.